Paget's Disease Family Risk Calculator
Estimate Your Risk
This tool estimates your likelihood of developing Paget's disease based on family history and genetic factors described in the article.
When bone pain or unexpected fractures pop up, many people blame old age or injury. But for a surprising number of cases, the culprit hides in the DNA. Paget's disease genetics play a crucial role in who gets the disorder and how severe it becomes. This guide walks you through the science, the key genes, testing options, and what you should do if a family member has been diagnosed.
What Is Paget's Disease?
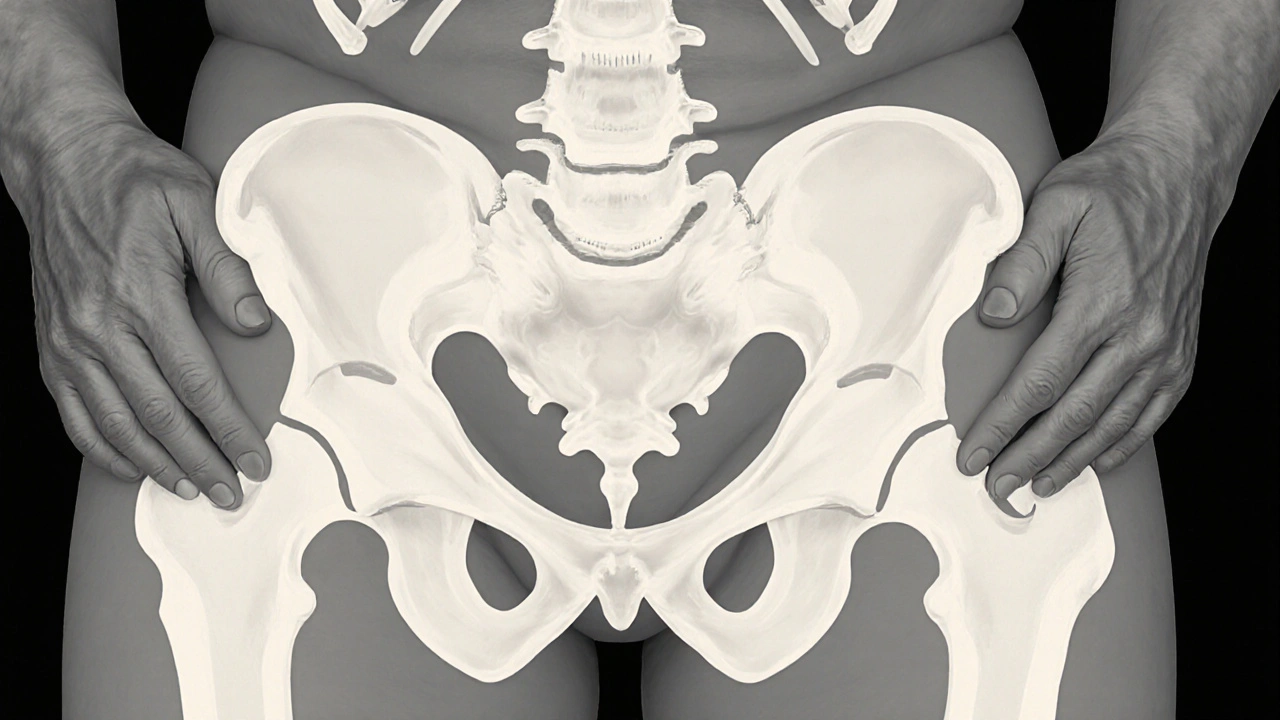
Paget's disease is a chronic bone disorder characterized by overly rapid bone remodeling, leading to enlarged, misshapen, and weak bones. It most commonly affects the pelvis, skull, spine, and long bones of the legs. Patients often experience bone pain, hearing loss (when the skull is involved), and, in advanced cases, fractures that occur with minimal trauma. The disease is more frequent in people over 50, but genetics can make it appear earlier.
How Genetics Shapes Bone Remodeling
Every bone cell follows a script written in our DNA. Genetics refers to the inherited instructions that govern cell behavior, including the activity of bone‑building (osteoblast) and bone‑resorbing (osteoclast) cells. When certain genetic variants disrupt the balance between these cells, the normal cycle of bone formation and breakdown goes haywire, setting the stage for Paget's disease.
Key Genes Linked to Paget's Disease
Research over the past two decades has pinpointed several genes that, when mutated, increase the risk of developing Paget's disease. The most influential is the SQSTM1 gene, which encodes the p62 protein involved in cellular signaling pathways that regulate osteoclast activity. Other notable genes include those in the RANK/RANKL/OPG axis, crucial for osteoclast differentiation.
| Gene | Typical Mutation | Effect on Bone Cells |
|---|---|---|
| SQSTM1 | P392L, P392Q (missense) | Hyperactive osteoclasts → accelerated bone resorption |
| TNFRSF11A (RANK) | Gain‑of‑function variants | Enhanced osteoclast formation |
| TNFRSF11B (OPG) | Loss‑of‑function variants | Reduced inhibition of RANKL → more osteoclast activity |
| VCP | Rare missense changes | Disrupted protein degradation, indirect osteoclast activation |
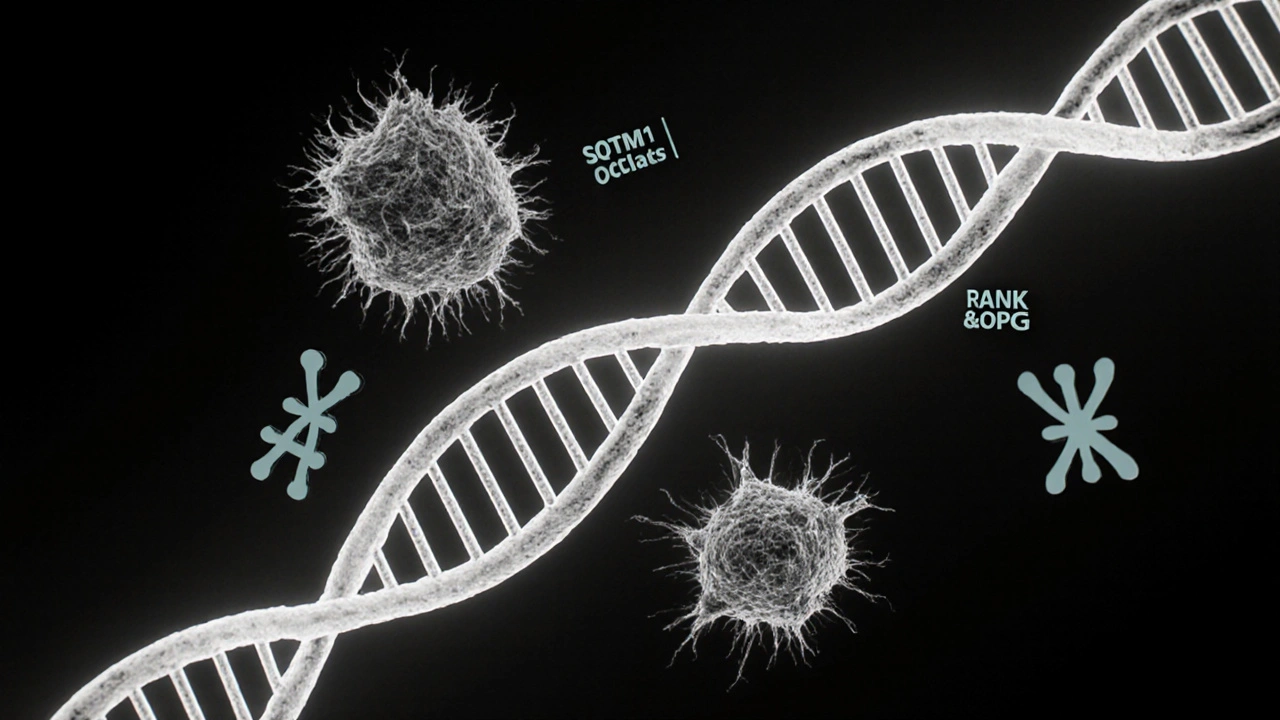
Inheritance Patterns and Family Risk
Paget's disease does not follow a classic Mendelian inheritance like cystic fibrosis. Instead, it shows a multifactorial pattern: a combination of genetic susceptibility and environmental triggers (such as viral infections). Yet families with multiple affected members often carry a familial Paget's disease form, typically linked to SQSTM1 mutations. If you have a first‑degree relative with Paget's, your risk jumps from the baseline 1‑2% to roughly 10‑15%.
Genetic Testing Options
When a clinician suspects a hereditary component, they may recommend genetic testing. The most common approach is targeted DNA sequencing, which looks specifically at the SQSTM1, RANK, and OPG genes. If the test is negative but suspicion remains, a broader whole‑exome sequencing can capture rare variants across the genome.
- Sample type: Blood or saliva.
- Turnaround time: 2‑4 weeks for targeted panels, 6‑8 weeks for whole‑exome.
- Cost (US, 2025): $350-$600 for targeted testing; $1,200-$1,800 for whole‑exome.
Results are interpreted by a genetic counselor who can explain the implications for you and your relatives.
Implications for Treatment
Knowing the genetic background can guide therapy. The mainstay treatment for Paget's disease is bisphosphonate therapy, which suppresses osteoclast activity and helps normalize bone turnover. Some studies suggest that patients with SQSTM1 mutations respond especially well to a single high‑dose infusion of zoledronic acid, achieving longer remission periods.
Beyond medication, lifestyle measures-adequate calcium and vitaminD, weight‑bearing exercise, and fall‑prevention strategies-remain essential regardless of genetic status.
Practical Steps for Patients and Families
- Take a detailed family history: Note any relatives with bone pain, fractures, or a formal Paget's diagnosis.
- Ask your doctor for a bone scan: Technetium‑99m bone scintigraphy can reveal early lesions before symptoms appear.
- Consider genetic counseling: If multiple family members are affected, a counselor can discuss testing and risk communication.
- Stay on top of monitoring: Serial alkaline phosphatase (ALP) blood tests help track disease activity after treatment.
- Adopt bone‑healthy habits: Regular weight‑bearing activity, balanced diet, and avoidance of smoking.
By combining family insight with modern genetic tools, patients can catch Paget's disease early, tailor therapy, and reduce complications.
Frequently Asked Questions
Can Paget's disease be inherited?
Yes, especially when a mutation in the SQSTM1 gene is present. While most cases are sporadic, families with multiple affected members often carry this hereditary variant, raising the risk for close relatives.
What symptoms should prompt a bone scan?
Persistent bone pain, unexplained enlargements of the head or pelvis, hearing loss, or a sudden fracture without a clear injury are all red flags. A bone scan can detect abnormal remodeling before X‑rays show changes.
Is genetic testing mandatory for diagnosis?
No. Diagnosis relies on clinical, radiographic, and biochemical findings. Genetic testing is optional and mainly useful for family‑planning, assessing risk in relatives, or guiding treatment choices.
Do people with the SQSTM1 mutation always develop the disease?
Not necessarily. Penetrance is incomplete-some carriers never show symptoms, while others develop severe disease. Additional factors like age, environment, and possibly viral exposure influence whether the disease manifests.
Can lifestyle changes reduce the impact of a genetic predisposition?
Yes. Maintaining adequate calcium and vitaminD, staying active, and avoiding smoking help keep bone turnover in check. While they don’t erase the genetic risk, they can delay onset and lessen symptom severity.

Genetics provides a roadmap for families navigating Paget's disease, highlighting who might benefit from early screening.
Understanding SQSTM1 and its impact can empower patients to discuss targeted testing with their physicians.
Adopting calcium‑rich foods and regular weight‑bearing exercise further supports bone health while genetic insights guide personalized therapy.
Staying proactive reduces the likelihood of severe complications.
Oh, another list of genes to memorize-because we all have time to study SQSTM1 in our free moments.
Maybe the real breakthrough will be a pill that makes bone remodeling less dramatic.
Sharing personal experiences with bisphosphonate treatment helps demystify the process for newcomers.
Many patients report that a single infusion of zoledronic acid can quiet the disease for years, easing pain and improving mobility.
Combining this with regular monitoring of alkaline phosphatase levels ensures that any flare‑ups are caught early.
Community support also plays a vital role in managing the emotional toll.
Neglecting bone‑healthy habits while boasting about a genetic predisposition borders on irresponsible self‑care.
Families should view genetic counseling not as an optional luxury but as a duty to protect future generations.
Skipping recommended calcium and vitamin D supplementation merely invites preventable fractures.
Everyone benefits when knowledge translates into concrete lifestyle changes.
Testing every relative is excessive.
The interplay between osteoclast signaling pathways and genomic variants constitutes a symphony of molecular choreography that, when disrupted, yields the pathological crescendo known as Paget's disease.
The chief among the conductors of this melody is the p62 protein, encoded by SQSTM1, whose structural domains orchestrate NF‑κB activation and autophagic flux.
Missense mutations such as P392L and P392Q perturb the ubiquitin‑binding region, thereby amplifying osteoclastogenesis beyond physiological limits.
Concurrently, aberrations in the RANK/RANKL/OPG axis-whether gain‑of‑function in TNFRSF11A or loss‑of‑function in TNFRSF11B-remove critical checkpoints that ordinarily temper bone resorption.
It is noteworthy that these genetic determinants rarely act in isolation; epistatic interactions modulate penetrance, rendering some carriers asymptomatic well into late adulthood.
Environmental variables, including latent viral exposures, may serve as catalysts that tip the balance toward overt disease in genetically predisposed individuals.
Whole‑exome sequencing, while comprehensive, often uncovers incidental variants whose clinical relevance remains ambiguous, underscoring the necessity for expert interpretation.
In contrast, targeted panels focusing on SQSTM1, RANK, and OPG provide a rapid, cost‑effective diagnostic avenue, particularly when a strong familial pattern is evident.
The therapeutic implication of identifying a SQSTM1 mutation extends beyond risk assessment; studies suggest heightened responsiveness to high‑dose zoledronic acid, potentially prolonging remission.
Nevertheless, clinicians must counsel patients that bisphosphonates constitute a symptomatic intervention, not a cure, and that vigilant follow‑up remains indispensable.
Lifestyle adjuncts-adequate calcium, vitamin D, and regular weight‑bearing activity-function synergistically with pharmacotherapy, mitigating the relentless cycle of bone turnover.
From a public‑health perspective, cascade screening of first‑degree relatives could curtail disease burden, yet ethical considerations demand respect for autonomy and informed consent.
Moreover, insurance coverage for genetic testing varies widely, prompting a need for advocacy to ensure equitable access.
As our genomic toolbox expands, future research may unmask additional modifiers, perhaps unveiling novel targets for precision medicine.
Until such breakthroughs materialize, a judicious blend of genetic insight, clinical vigilance, and patient‑centered care remains the optimal strategy for confronting Paget's disease.
That deep dive really clarifies how each gene fits into the bigger picture.
When you break it down like this, it’s easier for patients to see why testing matters.
Keep sharing these insights-they’re gold for the community.
While the stakes are high, it’s also okay to take things step by step.
Genetic counseling can feel daunting, but it’s just another tool in the toolbox.
Balance is key.
Sure, because memorizing gene names is the pinnacle of weekend fun.
Let’s hope the next breakthrough involves a pop‑quiz format.
Totally agree! 🎉 Sharing real‑world experiences makes the whole process less scary.
Keeping an eye on alkaline phosphatase is a game‑changer, and community stories give us hope.
Thanks for the reminder!
In our nation, the lack of universal genetic screening is a glaring oversight that betrays the very citizens it purports to protect.
When policymakers ignore the mounting evidence linking SQSTM1 variants to debilitating bone pathology, they prioritize cost‑saving over human welfare.
Such neglect fuels a cycle of preventable fractures, lost productivity, and needless suffering.
We must demand that our health system integrates comprehensive bone health assessments as a patriotic duty.
The analysis is thorough; however, the claim that “environmental variables…may serve as catalysts” requires citation.
Without solid references, the statement teeters on speculation.
Recent meta‑analyses indicate that carriers of the P392L SQSTM1 mutation exhibit a statistically significant increase in serum alkaline phosphatase levels compared with non‑carriers (p < 0.01).
This biochemical marker correlates with disease severity and can guide dosing intervals for bisphosphonate therapy.
Incorporating these data into clinical algorithms enhances precision medicine approaches for Paget's disease.
One might contemplate the dichotomy between genetic determinism and the agency of lifestyle, recognizing that the genome merely sketches a blueprint while habitual actions sculpt the edifice of health.
Thus, embracing both molecular insight and disciplined self‑care epitomizes a holistic philosophy toward disease mitigation.
Calling neglect “irresponsible” is an oversimplification; many patients lack access to affordable supplements and counseling due to systemic inequities.
Blaming individuals ignores the broader socioeconomic forces at play.
There are whispers that the pharmaceutical push for bisphosphonates masks undisclosed side‑effects, and that data suppression is coordinated through hidden channels.
Scrutinizing the source of research funding becomes essential when navigating treatment choices.