
When you see a new medication on the market that looks like an older, expensive drug but costs less, you might wonder if it is truly safe. For small-molecule drugs, we have generics. But for complex biological medicines, we have biosimilars, which are highly similar versions of already-approved reference biologics. Unlike generic pills, which are identical copies, biosimilars are not exact replicas. They are made in living cells, meaning tiny variations are natural. So, how does the U.S. Food and Drug Administration (FDA) decide if a biosimilar is good enough? The short answer is that they don't give them a letter grade or a star rating. Instead, they use a rigorous scientific evaluation process to determine if there are "no clinically meaningful differences" between the new product and the original.
The Legal Foundation: BPCIA and Section 351(k)
To understand how biosimilars are rated, you first need to know the rules of the game. The pathway for approving these drugs was created by the Biologics Price Competition and Innovation Act (BPCIA), enacted in 2009 as part of the Affordable Care Act. This law established an abbreviated licensure process under section 351(k) of the Public Health Service Act. Before this, every new biologic had to go through massive, costly clinical trials from scratch. The BPCIA allowed manufacturers to rely on the FDA's previous safety and efficacy data for the reference product, saving time and money while maintaining high safety standards.
The core requirement is simple in theory but complex in practice: the manufacturer must prove their product is "highly similar" to the reference product, despite minor differences in inactive components. As of late 2025, the FDA has approved 43 biosimilar products using this pathway, starting with Zarxio in 2015. These approvals have helped reduce costs for patients by approximately 15-30% compared to reference products, according to FDA economic analyses.
The "Totality of Evidence" Approach
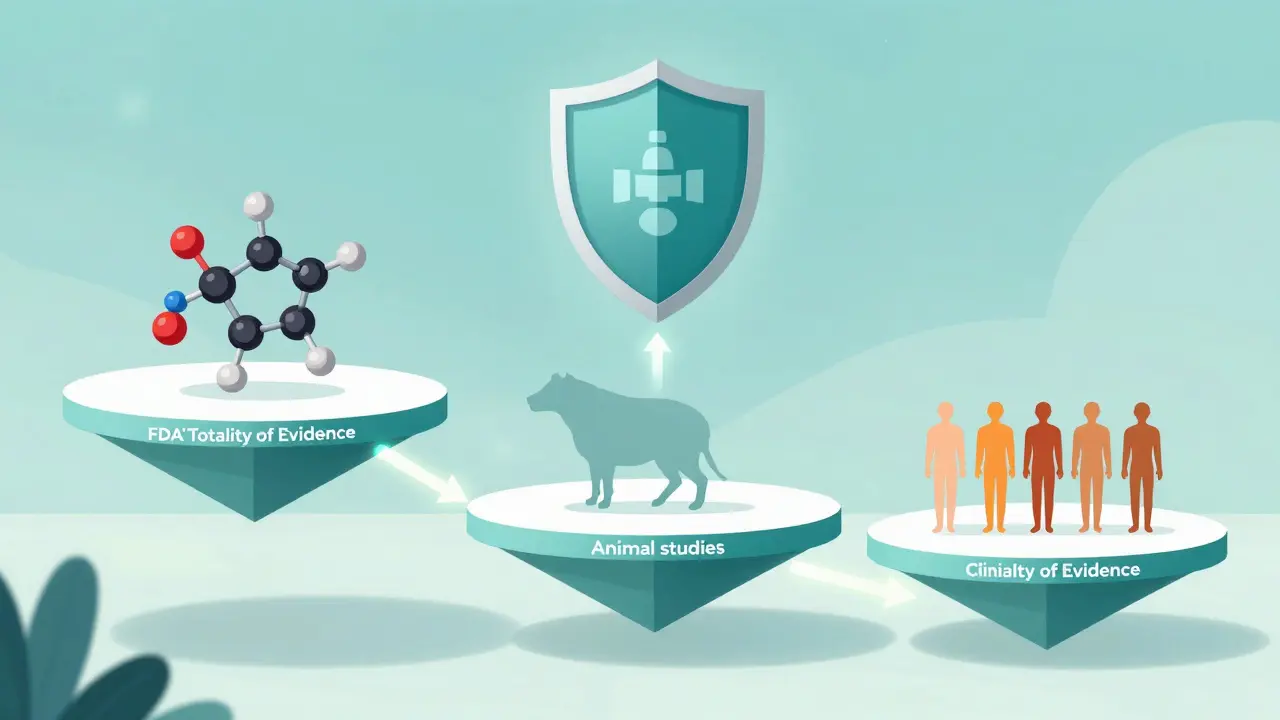
The FDA does not look at a single test result to approve a biosimilar. Instead, they use a "totality of evidence" approach. Think of it like building a case in court; one piece of evidence might be weak, but when you combine several strong pieces, the picture becomes clear. This process is stepwise, starting with the most detailed analysis possible before moving to animal or human studies.
- Analytical Studies: This is the foundation. Manufacturers use state-of-the-art techniques to characterize the molecular structure, purity, and biological activity of both the biosimilar and the reference product. The FDA evaluates up to 200-300 critical quality attributes. Similarity thresholds typically require 95-99% analytical similarity depending on the attribute. If the molecules look nearly identical under the microscope, the FDA may waive further testing.
- Animal Studies: If analytical data isn't enough, toxicity assessments in relevant species may be required. However, the FDA has discretion to skip this if the analytical data is robust.
- Clinical Studies: The final step usually involves pharmacokinetic (how the body absorbs the drug) and pharmacodynamic (what the drug does to the body) studies in humans. Immunogenicity assessment-checking if the body develops an immune response to the drug-is mandatory for all biosimilars.
A significant shift occurred in September 2024 when the FDA finalized updated guidance allowing manufacturers to potentially eliminate comparative efficacy studies in certain circumstances. If advanced analytical techniques provide sufficient evidence of biosimilarity, developers can skip some large-scale clinical trials, saving $50-100 million per product and accelerating timelines by 12-18 months.
Interchangeability: A Higher Standard
Not all biosimilars are created equal in the eyes of pharmacists. Some receive a designation called "interchangeable." This is a higher standard than basic biosimilarity. To earn this label, a product must demonstrate that it can be expected to produce the same clinical result as the reference product in any given patient. Furthermore, there must be no increased risk in terms of safety or efficacy when alternating or switching between the reference product and the biosimilar.
As of October 2025, only 17 of the 43 approved biosimilars hold this interchangeable status. This matters because state laws vary on whether pharmacists can automatically substitute a biosimilar for a reference product without the prescriber's consent. Interchangeable biosimilars generally face fewer barriers to substitution, making them more accessible to patients who want lower-cost options.
| Feature | Biosimilar | Interchangeable Biosimilar |
|---|---|---|
| Similarity Requirement | Highly similar to reference product | Highly similar + additional switching data |
| Clinical Meaningful Differences | None in safety, purity, potency | None, plus proven safety in switching |
| Pharmacist Substitution | Varies by state law | Easier in many states |
| Number Approved (as of Oct 2025) | 43 | 17 |
The Purple Book: Where to Find the Ratings
If you want to check the status of a specific biologic, you need to look at the Purple Book, the FDA's official listing of licensed biologics. Updated significantly in June 2021 pursuant to Section 325 of the Consolidated Appropriations Act, the Purple Book now includes mandatory patent information. Reference product sponsors must submit patent details within 30 days of disclosing them to biosimilar applicants. This transparency helps prevent legal surprises and allows developers to navigate the "patent thicket" that often delays market entry.
As of October 2025, the Purple Book lists 387 reference biologics and 43 approved biosimilars. It provides key data points such as licensure dates, exclusivity periods, and interchangeability status. In Q1 2025, the Purple Book transitioned to a fully searchable, API-accessible database with daily updates, replacing the slower monthly schedule. This makes it easier for healthcare providers and patients to verify the current regulatory standing of a medication.

Challenges and Real-World Performance
Despite the rigorous approval process, getting a biosimilar to market is hard. The median time from IND submission to approval in the U.S. is 3.2 years, compared to 2.1 years in the European Union. This delay is largely due to the FDA's more stringent analytical requirements and extensive patent litigation. While the FDA approved 43 biosimilars between 2015 and 2025, only 29 have actually launched, with an average delay of 11.3 months from approval to launch due to legal battles.
However, once they are on the market, do they work? Real-world evidence suggests yes. Data from the FDA's Sentinel Initiative shows that adverse event reports for biosimilars (0.8 per 10,000 patients) are statistically equivalent to reference products (0.7 per 10,000 patients) across all approved products as of Q3 2025. Patient advocacy groups, including the Cancer Support Community, have praised the FDA's stepwise evaluation, noting that no biosimilar-specific safety signals have been identified in nine years of post-marketing surveillance.
Future Outlook: AI and Complex Molecules
The landscape is evolving rapidly. The FDA's 2025-2027 Biosimilars Roadmap prioritizes developing guidance for complex molecules like antibody-drug conjugates and gene therapies, which currently lack standardized assessment methods. Additionally, the agency plans to implement artificial intelligence tools for analytical data review in a pilot program launching in Q1 2026. These changes aim to increase annual approvals from 3-5 to 7-9 by 2027, potentially reducing development timelines from 6.8 years to 5.2 years. As the technology advances, the definition of "similarity" will continue to refine, ensuring that patients benefit from innovation without compromising safety.
Are biosimilars exactly the same as the original biologic?
No, biosimilars are not identical copies like generic small-molecule drugs. Because biologics are produced in living cells, minor variations are natural. However, biosimilars must be "highly similar" to the reference product with no clinically meaningful differences in safety, purity, or potency.
What does it mean for a biosimilar to be "interchangeable"?
Interchangeability is a higher regulatory standard. It means the biosimilar can be expected to produce the same clinical result as the reference product in any patient, and there is no increased risk when switching between them. This designation often allows pharmacists to substitute the biosimilar for the reference product without prior prescriber authorization, depending on state laws.
How long does it take for the FDA to approve a biosimilar?
The median time from Investigational New Drug (IND) submission to approval in the U.S. is approximately 3.2 years. This is longer than in the EU (2.1 years) due to the FDA's rigorous analytical requirements and frequent patent litigation delays.
Where can I find a list of approved biosimilars?
You can find the official list in the FDA's Purple Book. As of 2025, it contains 43 approved biosimilars and 387 reference biologics. The book includes details on licensure dates, patent information, and interchangeability status.
Is it safe to switch from a reference biologic to a biosimilar?
Yes, extensive real-world data supports the safety of switching. The FDA's Sentinel Initiative found that adverse event rates for biosimilars are statistically equivalent to reference products. No biosimilar-specific safety signals have been identified in over nine years of post-marketing surveillance.
